By Sierra Pellechio, Hepatitis Delta Connect Coordinator
Hepatitis delta is an aggressive form of hepatitis that can only exist alongside hepatitis B. This means that all hepatitis B patients are at risk for hepatitis delta, but so are people who have not received the hepatitis B vaccination series.
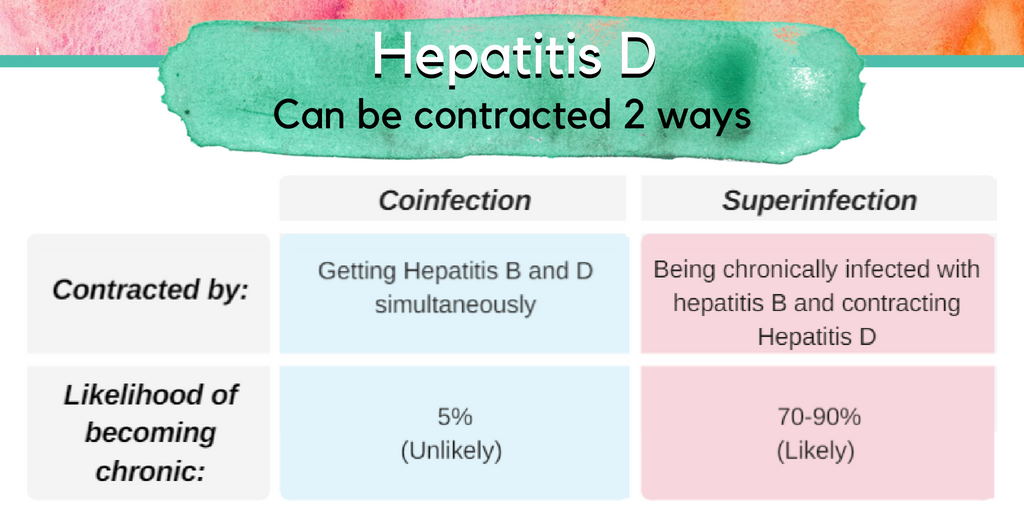
If contracted, 70-90% of people with chronic hepatitis B will go on to also develop a chronic hepatitis delta infection – called a “superinfection”. Approximately 70% of these cases will progress to cirrhosis (liver scarring), compared to 15-30% of those infected only with the hepatitis B virus.
Due to the likelihood of liver complications, hepatitis B patients should be aware of potential exposures to hepatitis delta. The virus is spread the same way as hepatitis B, through direct blood-to-blood contact and unprotected sex with an infected person. It is important to be aware that blood contact could also occur by exposure to unsafe blood transfusions, unsterile medical or dental equipment, and the sharing of razors or toothbrushes with an infected person due to the possibility of infected blood entering the body.
People who are not infected with hepatitis B may be at risk for “coinfection”, when someone contracts hepatitis B and delta simultaneously during one exposure. In these cases, greater than 90% of adults will clear both infections and develop protective antibodies. While a co-infection generally resolves spontaneously after about 6 months, it can sometimes result in a life-threatening or fatal liver failure.
The good news is that the hepatitis B vaccine series can prevent both viruses in people who are not already infected. Once completed, the vaccine can provide a lifetime of protection!
Have you recently been told you have hepatitis B? Dealing with the diagnosis and waiting out the next six months to determine if your infection will resolve itself or learning that it is a chronic infection can be nerve-wracking.
Fortunately, greater than 90 percent of healthy adults who are newly infected will clear or resolve an acute hepatitis B infection. On the hand, greater than 90% of babies and up to 50% of children infected with hepatitis B will have lifelong, chronic infection. Sometimes people are surprised to learn they have a chronic infection. It can be confusing since there are typically few or no symptoms for decades. If a person continues to test hepatitis B positive for longer than 6 months, then it is considered a chronic infection. Repeat testing is the only way to know for sure.
Acute hepatitis B patients rarely require hospitalization, or even medication. If you are symptomatic, (some symptoms include jaundice, dark urine, abdominal pain, fever, general malaise) you may be anxiously conferring with your doctor, but if you are asymptomatic, you might not feel compelled to take the diagnosis seriously. Ignoring your diagnosis can be very serious. If you have concerning symptoms like jaundice (yellow eyes and skin), a bloated abdomen or severe nausea and vomiting, please see your doctor immediately. Your doctor will be monitoring your blood work over the next few months to see if you clear the virus, or monitoring your liver if there are concerning symptoms.
Your job is to start loving your liver …today. STOP drinking alcoholic beverages. Refrain from smoking cigarettes. Your liver is a non-complaining organ, but you cannot live without it. Make your diet liver-friendly and healthy filled with a rainbow of vegetables and fruits, whole grains, fish and lean meats. Minimize processed foods, saturated fats and sugar. Drink plenty of water.
Talk to your doctor before taking prescription medications, herbal remedies, supplements or over-the-counter drugs. Some can be dangerous to a liver that is battling hepatitis B. Get plenty of rest, and exercise if you are able.
Don’t forget that you are infectious during this time, and that loved ones, sexual partners and household contacts should be tested to see if they need to be vaccinated to protect against hepatitis B. Sometimes family members or close household contacts may find that they have a current infection or have recovered from a past HBV infection. If anyone fears exposure, ensure them that hepatitis B is not transmitted casually. They should get tested, and vaccinated if needed, and take simple precautions. Remind them that 1/3 of the world’s population will be infected with the hepatitis B virus during their lifetime.
On the flip-side… Do not let this new hepatitis B diagnosis consume you. As the weeks and months pass, you might find that the infection is not resolving, and you might worry that you have a chronic infection. The associated stress and anxiety can be challenging, even overwhelming. It can contribute to physical symptoms you may be experiencing. Find a family member, friend, or health care professional with whom you can share your concerns.
If you are told you have recovered from an acute HBV infection (you are now HBsAg negative, HBcAb positive and HBsAb positive) be sure to get copies of your lab reports to ensure there are no mistakes. Compare them with our easy to use blood tests chart. If something looks wrong, or if you’re confused, speak up and ask your doctor. Once confirmed, be sure to include hepatitis B as part of your personal health history. This is important in case you have conditions requiring treatment later in life that might once again warrant monitoring of your hepatitis B. It is possible for a past HBV infection to reactivate if a person requires longterm immune suppressing drugs .
No one wants to learn they have chronic hepatitis B but it is a manageable disease. You’ll want to see a doctor with experience treating chronic HBV so they can run additional tests. There are very effective treatments available, though not everyone with chronic HBV needs treatment. All people living with chronic HBV benefit from regular monitoring since things can change with time. Please do not panic or ignore a chronic hepatitis B diagnosis. Take a deep breath and get started today learning more about your HBV infection and the health of your liver. Things are going to be okay!
If you are confused about your diagnosis, please feel free to contact the Hepatitis B Foundation at info@hepb.org.
Welcome to “Journey to the Cure.” This is a web series that chronicles the progress at the Hepatitis B Foundation and Baruch S. Blumberg Institute towards finding the cure for hepatitis B.
In the third episode (part 2), Kristine Alarcon, MPH sits down with Maureen Kamischke, Social Media Manager for the Hepatitis B Foundation, to talk about her social media work at the Foundation. For any questions about hepatitis B, please email info@hepb.org.
Disclaimer: The information provided in this audio post is not intended to serve as medical advice of endorsement of any product. The Hepatitis B Foundation strongly recommends each person discuss this information and their questions with a qualified health care provider.
Edited by:
Kristine Alarcon, MPH
Special thanks:
Samantha Young
Music:
Modern – iMovie Library Collection
Script:
Welcome to “Journey to the Cure!” Every month, we’ll sit down with scientists from the Hepatitis B Foundationand the Baruch S. Blumberg Institute to talk to you about hepatitis B and efforts to find a cure for hepatitis B. There’s still a long way to go, but we’re here to walk you through our journey.
Kristine Alarcon, MPH:
You are our social media manager, and I know you have also shared your hepatitis B story. You can find Maureen’s story in our #justB campaign. But, can you tell me more about your work as a social media manager?
Maureen Kamischke: At the Foundation, we are very active on three outlets: Twitter, Facebook, and Instagram. On Twitter, we have over 6,000 followers. We have a very active Facebook community. I would really encourage people to check out these outlets. It’s a great place to just check out what’s going on: drugs and the status of them on a daily basis. Basically, those are being updated every day.
Kristine Alarcon, MPH:
So, why is social media so important to conveying hepatitis B information?
Maureen Kamischke: So, I think social media is a great way to reach out to different audiences. I think it’s a great way to get the messages out. You know, you can put messages out; you can link back to different parts of our website that really need to be featured and highlighted so that there are areas of what people want to learn more about; and then of course, if you are really interested in the most recent articles in hepatitis B, it’s an easy enough to link out to those so that you are not doing the work for it.
Kristine Alarcon, MPH:
So, it’s just like another type of way to easily disseminate information and get it more widely available to everyone.
Maureen Kamischke: Yes.
Kristine Alarcon, MPH:
So, you’ve made so many connections across the globe in regard to hepatitis B partnerships, so what do you think the future looks like in the elimination of hepatitis B?
Maureen Kamischke: Well, I would have to say that on behalf of myself and all of our friends around the world, we’re all waiting for the cure, but until that time, there’s a lot that we can do. We have a lot of good treatments available. There’s a lot of information that needs to be disseminated. There are a lot of issues with stigma and discrimination. And hopefully, social media can help decrease the amount of stigma and discrimination by educating people, allowing them to learn more about the disease, more about the people that are living with it. It’s devastating the impact of the disease that it has on people, and this is a great way to reach out to them.
Kristine Alarcon, MPH:
Thank you so much for all your efforts. Be sure to join us on our next episode of “Journey to the Cure.” Just wanted to thank Maureen again for all of her time and all of her efforts in conveying such wonderful information around the world.
I thought hepatitis B was sexually transmitted? I just tested positive, but my partner tested negative, we’ve been together for years, what gives?
This question is a common one. Hepatitis B can be transmitted sexually, so why do some people — who were not vaccinated — never get hepatitis B from their sexual partners?
It comes down to factors, such as the type of sexual activity partners engage in, the viral load (HBV DNA) of the infected partner, and who is on the receiving end of infectious body fluids, especially blood (which contains the most virus), and semen.
Having one partner infected, while the other is not, can add more stress to an already traumatic hepatitis B diagnosis. “It was very confusing and made me question how was it possible I was the only one infected,” said a woman who tested positive while her husband tested negative. “I thought it was possibly a mistake, maybe I was a biological anomaly, which of course I was not.”
Let’s look at the factors that may play a role in transmission of hepatitis B infection through sexual activity.
Viral load: Semen, vaginal fluids and blood all contain the hepatitis B virus (HBV), and the higher the viral load in the blood of an infected individual, the more infectious they are considered to be. Having an undetectable viral load might reduce or eliminate the chance of transmitting the virus to someone during unprotected sex; research is still trying to assess whether a person with an undetectable viral load in the blood is able to transmit the virus through sex. This is a good reason for individuals living with hepatitis B to talk to their doctor about the benefits of starting antivirals if they have detectable HBV viral load in their blood; treatment which lowers the viral load in the blood might also serve as a prevention measure for transmitting the virus.
Once an individual tests positive for hepatitis B surface antigen (HBsAg), they should encourage their partners to get screened for hepatitis B, and vaccinated if they are still susceptible to the virus.
The timing of sexual activity: An infected person who is menstruating is more likely to transmit hepatitis B infection to an unvaccinated partner, because menstrual blood can contain higher levels of HBV than vaginal secretions. That is why dental dams and condoms are recommended to provide a reasonable barrier against exposure, during that time of the month.
The type of sexual activity: Certain sexual activities are far more efficient at transmitting hepatitis B virus than others. Oral sex appears to have a lower rate of hepatitis B transmission than vaginal sex. Anal sex carries a higher risk of transmission because of tears in the skin that can occur during penetration, which increases the likelihood of transmission of HBV to an unvaccinated partner.
Fingering carries a lesser risk, unless the infected partner is menstruating while the other partner has bruises or cuts on their hands that could allow entry of hepatitis B virus from the body fluid into the bloodstream. In such cases, gloves are highly recommended.
The hepatitis B status of the other partner: The “uninfected” partner could have already been infected and cleared the virus, or vaccinated as an infant. When a person is first diagnosed with hepatitis B, doctors often test his or her partner for only the hepatitis B surface antigen (HBsAg), which indicates a current hepatitis B infection. If they are negative for HBsAg, they are advised to receive the hepatitis B vaccine as soon as possible. However, this does not mean that they were never infected.
Testing for the hepatitis B surface antibody (also known as anti-HBs or HBsAb), and hepatitis B core antibody (HBcAb) is the only way to identify a past recovered infection or prior vaccination.
Hepatitis B is often called the “silent” infection because many people who get hepatitis B may not experience any of the alarming symptoms (like fever or jaundice). As a result, many individuals may never realize they were infected. A partner who tested negative for HBsAg, may actually have been infected in the past and cleared the infection and now has protective hepatitis B surface antibodies to forever safeguard them from infection. If they’re vaccinated without proper screening, then tested for HBsAb after vaccination, they will test positive for surface antibodies, without ever knowing that their antibodies resulted from a past infection, not immunization.
Bottom line, if one partner is diagnosed with hepatitis B and the other is not, it might seem unusual, but it is not uncommon. Just like any other virus, there is not a 100% chance of transmission with exposure. The undiagnosed partner should get tested using the 3-panel blood test (HBsAg, HBsAb, and HBcAb) and immediately vaccinated if they are still vulnerable to a hepatitis B infection (HBsAb negative).
The is safe, effective, and provides lifelong protection.
Take a quiz to find out how much you know about hepatitis B transmission: click here.
Tell us why you think hepatitis B testing is important?
Globally, 292 million people are living with chronic hepatitis B. Only 10 percent are aware of their diagnosis. The theme for this year’s World Hepatitis Day is “Find the Missing Millions.” Help us raise awareness for World Hepatitis Day (July 28th, 2018) by telling the world why it is important to get tested for hepatitis B!
Create an awareness message about hepatitis B by answering the prompt below.The Hepatitis B Foundation will compile video entries for a larger video that will be released on World Hepatitis Day, July 28, 2018.
Who Can Enter? Anyone across the world!
Here’s how to Enter:
Record a short video or an audio clip of yourself (15 seconds or less) answering the prompt, “People should be tested for hepatitis B because ….”
2. Note: You may choose the audio option if you wish to remain anonymous. Film yourself answering the above question. Your face and/or your picture does not have to be in the video; however, we must be able to hear you. If you choose to record an audio clip you are welcome to send a picture from your country or something that represents you.
Keep your video no longer than 15 seconds!
Send your video to us:
By uploading your video to our Google form(You must have a gmail account)
You can also email the file using wetransfer. Please ensure that you send the email to info@hepb.org.
When you send your video, please mention that you wish to participate in the World Hepatitis Day 2018 Campaign.
Video Tips/Guidelines
Your video must be 15 seconds or less
Your video should be in English
Note: If your video is recorded in a language other than English, please provide the English translation. If possible, provide a timed script with timings of phrases.
Videos must be recorded in Landscape/horizontal mode. Videos recorded in a Vertical format cannot be used.
Record your video in a quiet area or with a microphone.
Record your video in good lighting.
Disclaimer
By submitting a video to this campaign, participants give the Hepatitis B Foundation permission to use their videos (audio and video), in the World Hepatitis Day campaign and promotion, as well as in future hepatitis B awareness efforts. The participant will waive any claims to royalty, right, or remuneration for such use. The Hepatitis B Foundation will not disclose any personal information obtained from participants (i.e., full names, email addresses, etc.) in the campaign to third parties or use the information for marketing or other purposes.
Welcome to “Journey to the Cure.” This is a web series that chronicles the progress at the Hepatitis B Foundation and Baruch S. Blumberg Institute towards finding the cure for hepatitis B.
In the third episode (part 1), Kristine Alarcon, MPH sits down with Maureen Kamischke, Hepatitis B Foundation Social Media Manager, to discuss what expectant mothers can do when they have hepatitis B.
For any questions about hepatitis B, please email info@hepb.org.
The Hepatitis B Foundation is a national nonprofit organization dedicated to finding a cure and improving the lives of those affected by hepatitis B worldwide through research, education and patient advocacy. Visit us at www.hepb.org, on Facebook at www.facebook.com/hepbfoundation, on Twitter at @hepbfoundation, and our Blog at www.hepb.org/blog
Disclaimer: The information provided in this video is not intended to serve as medical advice or endorsement of any product. The Hepatitis B Foundation strongly recommends each person discuss this information and their questions with a qualified health care provider.
Although there are highly effective treatments available to manage hepatitis B, there are few available treatments for hepatitis D, and none are U.S. Food and Drug Administration (FDA) approved. Hepatitis D is the most severe form of viral hepatitis, and coinfection can accelerate liver damage and cause cirrhosis or liver cancer in as little as 5 years for some patients. Currently there is no approved drug for acute or chronic hepatitis B/D coinfection, but in trials pegylated interferon alpha has shown to be somewhat effective. By stimulating the body’s immune system, around 25-30% of patients are able to suppress their hepatitis D viral load with weekly injections over 48 weeks. Emerging research is showing higher rates of effectiveness with prolonged interferon treatment beyond one year, but it can be difficult for patients to continue due to the physical and mental toll of interferon on the body. Antiviral medications that are proven effective against hepatitis B are sometimes prescribed along with interferon therapy for patients with a high hepatitis B viral load, but these have no effect on hepatitis D. It is urgent that more treatment options be developed for the millions of hepatitis B/D patients that are eagerly awaiting them.
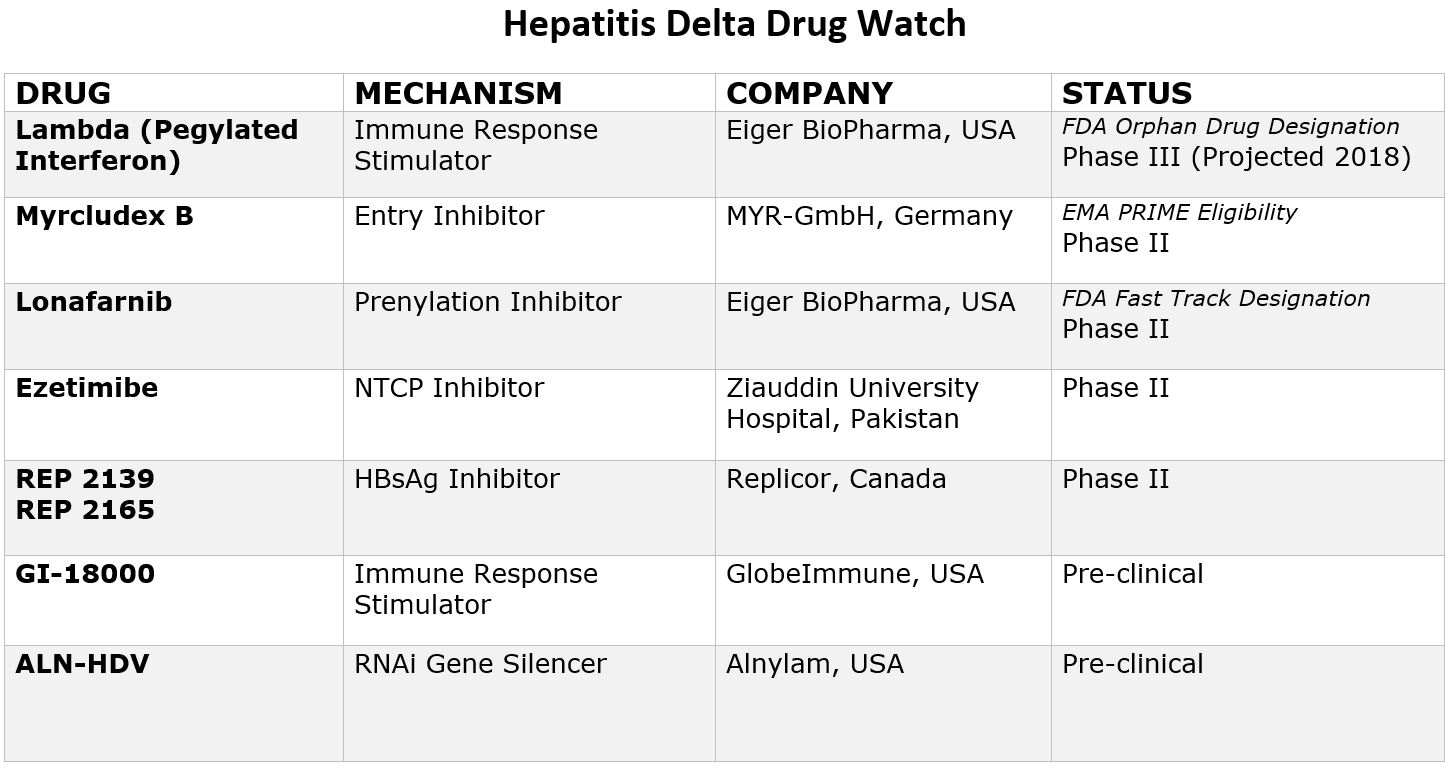
The good news is that with renewed scientific interest, research and funding, eight new drugs are currently in development that offer hope for more treatment options in the coming years. Two drugs have even been granted special designations by the FDA and one by European Medicines Agency (EMA), paving the way for increased resources and funding for development. Due to recent advancements, the future looks hopeful, and within a few years it is likely there will be more treatment options available. Below is a chart that provides more information on these new drugs and their current clinical trial status.
Pegylated Interferon Lambda
Pegylated-interferon-lambda (PEG-IFN-λ) is a well-characterized, late-stage, first in class, type III interferon that stimulates cell-mediated immune responses that are critical for the development of host protection during viral infections. This drug has now been granted “Orphan Drug Designation” by the FDA, fast-tracking the development process.
Myrcludex B
This drug is an “entry inhibitor” that prevents the virus from entering into hepatocytes (liver cells) and has shown activity against the hepatitis B virus. It may also stop the development of a hepatitis D infection. A recent study showed promise for Myrcludex B when combined with PEG-INF in reducing hepatitis D viral levels. It has been granted PRIME Eligibility by the European Medicines Agency, a status that promotes support in development of drugs that serve an unmet medical need.
Ezetimibe
Currently used to lower cholesterol in the blood, Ezetimibe is being studied for effectiveness against hepatitis D. Ezetimibe possesses pharmacophore features to stop NTCP, the receptor required for hepatitis B and hepatitis D hepatocyte entry.
Lonafarnib
This drug works by targeting the protein assembly process, preventing the production of new virus particles. In a current clinical trial, Lonafarnib combined with Ritonavir has shown promise in reducing hepatitis D viral levels, and the FDA has granted it fast-track status since this class of drugs have been developed for the treatment of cancers and have been shown to be safe.
Rep 2139
This compound is known as a “Nucleic acid-based Amphipathic Polymer” (NAP) which prevents the release of hepatitis B surface antigen (HBsAg) from infected liver cells and is being evaluated for hepatitis D virus in combination with pegylated interferon (PEG IFN).
GI-18000
GI-18000 Tarmogen is being studied for its effectiveness in causing a T cell immune response against cells infected with Hepatitis D and thereby improving outcomes. The strategy is to identify molecular targets that distinguish diseased cells from normal cells and activate the immune system to selectively target and eliminate only the diseased cells.
ALN-HDV
This approach is being used for both the hepatitis B and hepatitis D virus to “silence” the viral RNA with compounds that interfere with and cause the destruction of the viral genome (e.g. stop replication of the virus).
As clinical trials progress, sites may open across the world that are enrolling hepatitis D patients. Keep checking here for an up-to-date list of all current clinical trials.
Click here for more information about the phases of the clinical trial process.
Join Hepatitis B Foundation, NASTAD and CDC’s Division of Viral Hepatitis for a Twitter #HepChat at 2 p.m. (EST) Thursday, June 14. The chat will highlight Hepatitis Awareness Month outreach events and allow hepatitis B and C partner organizations to share their successes, challenges and lessons learned from their efforts. HBF’s Kristine Alarcon and Jason Crum, this month’s featured storyteller will also be LIVE on Facebook, so if you’re not on twitter join us at hepbfoundation.
May is Hepatitis Awareness Month and Saturday, May 19this National Hepatitis Testing Day in the United States. This day is an opportunity to increase awareness and testing for both hepatitis B and C. It is also a reminder for health care providers and the public of the importance of testing for viral hepatitis.
Why is hepatitis B testing necessary? Hepatitis B is largely asymptomatic, which means that symptoms don’t always occur or are not obvious. Some people will not know that they have hepatitis B until it is too late, or they may learn of their infection from a blood donation screening or lab work. There are groups of people who have a greater risk of hepatitis B compared to others, so it doesn’t hurt to be sure. here are some places around the world that have an extremely high hepatitis B prevalence (where many people are infected). It is important that people who are at high risk for a hepatitis B infection see a doctor to get tested, to find out if they have a hepatitis B infection. People living with chronic hepatitis B should be monitored regularly and appropriately screened for liver cancer. So, if you find you do have hepatitis B, talk to your doctor about what to do next.
Remember, hepatitis B does not discriminate. Don’t wait for symptoms. B sure. B tested. If you do not have hepatitis B, then give yourself lifelong protection with the hepatitis B vaccine. The hepatitis B vaccine is safe and effective. Children or adults can get the 3-shot vaccine series, and there isa newly approved two-dose adult vaccine to protect us against hepatitis B! However, the vaccine doesn’t work if you are already infected.
Don’t forget to check out these free, confidential hepatitis screenings this weekend! Check out Hep B United’s resource to find local events in your area. You can also visit the CDC’s website for more ideas on how to increase awareness on National Hepatitis Testing Day, and every day!
Hepatitis Victoria in Melbourne, Australia recently released their “bright, colorful, positive and silly” children’s book, Little Hep B Hero! In addition to creating a book, Hepatitis Victoria also created an animation.
Little Hep B Hero, which is available in English, Simplified Chinese and Vietnamese, gives children and their families a delightful glimpse into a young girl’s visit to her neighbor and friend, Rosa. As Rosa and the young girl prepare food and lemonade together, Rosa tells her about living with chronic hepatitis B. Rosa explains what the liver does, what hepatitis B is, how the virus is transmitted, how she maintains a healthy lifestyle with chronic hepatitis B, and shares tips on preventing hepatitis B transmission. Little Hep B Hero also provides cute and easy to understand visuals of the little girl as a superhero while Rosa explains this information.
The book does an excellent job of explaining liver functions and what the hepatitis B virus does to the liver! The analogy of using a sieve to demonstrate how the liver filters toxins in the body was creative. The book also does a great job of indirectly tackling some myths associated with hepatitis B. For example, people are often hesitant to share meals with those living with chronic hepatitis B. Little Hep B Hero lets its audience know that you cannot get hepatitis B through food preparation or sharing a meal, so Rosa cooks a meal for her neighbors.
Little Hep B Hero is an important read for future generations and their families! Getting the conversation started early about hepatitis B will address the stigma and discrimination associated with it. When children are talking about hepatitis B and are knowledgeable about it, hepatitis B isn’t a scary topic anymore. The message of hepatitis B as a family matter highlights the importance of educating and testing family members.
For more information, check out their press release, and to purchase Little Hep B Hero, visit Hepatitis Victoria’s website here.

 By Sierra Pellechio, Hepatitis Delta Connect Coordinator
By Sierra Pellechio, Hepatitis Delta Connect Coordinator